Молдова
Население Молдовы составляет около 3,5 млн. человек. Рынок медицинской и фармацевтической продукции на более чем 80% обеспечивается за счет импорта. Объем рынка лекарственных средств и медицинских изделий ежегодно растет. В 2016-2017 году правительство Молдовы предприняло ряд действий, направленных на увеличение количества реимбурсируемых лекарственных средств, привлечение международных производителей и улучшению конкуренции на фармацевтическом рынке, а также расширяет сети аптек в сельской местности.
Регуляторная система Республики Молдовы является независимой, однако законодательство в отношении медицинской продукции находится в активной гармонизации с законодательством Европейского Союза. Упрощенные процедуры регистрации предусмотрены для медицинских продуктов, получивших авторизацию на рынке ЕС и других стран с высокими регуляторными требованиями.
Для допуска на рынок Молдовы лекарственных средств необходимо провести государственную регистрацию. Все медицинские изделия подлежат государственной регистрации, основанной на процедурах оценки соответствия, до их размещения на рынке Молдовы. Специальные пищевые продукты проходят процедуру нотификации либо регистрации, и должны соответствовать требованиям Санитарного регламента о добавках к пище. Импортируемые косметические продукты проходят сертификацию в процессе таможенного оформления.
Лекарственные средства
Основными законодательными актами, регламентирующими процедуру регистрации и обращения лекарственных средств в Молдове являются:
- Закон Республики Молдова №1456-XII от 25.05.1993 «О фармацевтической деятельности»;
- Закон Республики Молдова №1409-XIII от 17.12.1997 «О лекарствах»;
- Приказ Министерства Здравоохранения Республики Молдова №739 от 23.07.2012 «об авторизации лекарственных средств для человека и утверждении пострегистрационных изменений».
Регистрацию лекарственных средств координирует Министерство здравоохранения Республики Молдова, а экспертизу регистрационных материалов проводит Агентство по лекарственным и медицинским изделиям (https://msmps.gov.md/ru/).
Заявитель регистрации – лицо, назначенное и уполномоченное владельцем в качестве его представителя в ходе проведения процедуры авторизации лекарственных средств в Республике Молдова. Владелец регистрационного свидетельства лекарственного средства – изобретатель, производитель или любое другое юридическое лицо, уполномоченное ими, ответственное за эффективность, качество и безопасность лекарственного средства. Заявителем регистрации может быть только резидент Республики Молдова, в то время как владельцем регистрации может быть как резидент, так и нерезидент.
Владелец регистрационного свидетельства несет ответственность за качество, эффективность и безопасность лекарственного средства, размещенного на рынке, и его соответствие требованиям Спецификации.
Лекарственное средство, подаваемое на первичную авторизацию в Республике Молдова, должно быть зарегистрировано либо в стране производства, либо в стране владельца регистрационного свидетельства, либо в одной из стран Европейского Экономического Пространства, Швейцарии, США, Канаде, Японии, Австралии.
Процедура регистрации состоит из следующих этапов:
- Заявитель подает в Агентство по лекарствам и медицинским изделиям формуляр заявления, а также — регистрационное досье, образцы лекарственного средства и референтные стандарты.
- Агентство по лекарствам и медицинским изделиям выставляет Заявителю счета для оплаты суммы за регистрацию.
- После поступления оплаты проводится первичная экспертиза: проверяется комплектность досье. При положительном результате первичной экспертизы заявление валидируется, при отрицательном результате — досье возвращается заявителю.
- Валидированное заявление на регистрацию и документация для регистрации распределяются группе экспертов и при необходимости, лаборатории по контролю качества лекарств.
- Проводится специализированная экспертиза регистрационного досье.
- Лаборатория по контролю качества лекарств проводит апробацию образцов лекарственного средства согласно Спецификации и методам контроля.
- В ходе специализированной экспертизы и/или лабораторного анализа образцов могут возникнуть замечания, на которые Заявителю необходимо предоставить полный ответ в течении не более 90 дней. Срок может быть увеличен письменным заявлением заявителя не более чем на 30 дней.
- В процессе экспертизы может быть принято решение о проведении проведения проверки производства, мест проведения доклинических/клинических испытаний, а также проверку у владельца регистрационного свидетельства или его представительства в отношении соответствия системы фармаконадзора. На время такой проверки (инспекции) процедура регистрации приостанавливается, но не более чем на 12 месяцев.
- Положительные отчеты по специализированной экспертизе и лабораторному контролю качества представляются Комиссии по лекарствам, которая принимает решение об утверждении лекарственных средств к регистрации.
- Агентство по лекарствам и медицинским изделиям составляет проект приказа об авторизации лекарственных средств, который затем утверждается Министерством здравоохранения.
- Лекарственные средства, внесенные в приказ Министерства здравоохранения, считаются включенными в Государственный регистр лекарств, разрешенных в Республике Молдова, со дня подписания соответствующего приказа.
- После подписания приказа выдается оригинал регистрационного свидетельства.
Срок специализированной экспертизы составляет до 210 дней. Однако данный срок не включает в себя (а) действия, которые выполняются до начала экспертизы: получение и оплата счетов, первичная экспертиза, (б) время, необходимое заявителю для подготовки ответов на замечания, (в) время, необходимое для проведения проверки производства, мест проведения доклиники/клиники, места ведения фармаконадзора, (г) действия, которые выполняются после принятия решения об утверждении лекарственного средства к регистрации: составление проекта Приказа и его подписание, включение лекарства в реестр (регистр), выдача оригинала свидетельства.
Заявитель имеет право приостановить процесс авторизации на срок до 5 лет.
Лабораторный контроль качества не проводится:
- для лекарственных средств, зарегистрированных EMA или произведенных в одной из стран Европейского экономического пространства или в Швейцарии, США, Канаде, Японии, Австралии;
- для лекарственных средств, место производства которых было инспектировано одной из стран-участниц/членом PIC/S, подтверждено сертификатом GMP, выданным уполномоченным органом этой страны.
Срок действия регистрационного свидетельства составляет 5 лет. Лекарственное средство может использоваться в медицинской практике до истечения срока годности.
Регистрационное свидетельство может быть аннулировано Министерством здравоохранения в случае, если лекарственное средство не поставлялось в страну в течение трех (3) лет с момента выдачи регистрационного удостоверения.
Упрощенная процедура регистрации
Упрощенная процедура регистрации предусмотрена законодательством Молдовы для лекарственных средств, зарегистрированных EMA как минимум в одной из стран Европейского экономического пространства или в Швейцарии, США, Канаде, Японии, Австралии.
Очевидными преимуществами упрощенной процедуры является уменьшенный срок экспертизы до 60 дней, отсутствие необходимости предоставления образцов и стандартов, отсутствие лабораторного контроля качества.
Упрощенная процедура регистрации действует при условии полной идентичности подаваемого досье последнему (актуальному) принятому досье в одной из указанных стран. При наличии досье, специфичного для рынка или региона препарат должен подаваться по стандартной процедуре.
Требования к регистрационному досье
Регистрационное досье подается в формате CTD:
- Модуль 1 подается в бумажном формате. Часть административной документации подлежит легализации. Eudra сертификаты разрешено подавать без нотариального заверения.
- Модули 2-5 подаются на на электронном носителе.
Документация подается либо на румынском, либо на английском, либо на русском языке.
SmPC подается в переводе на румынский язык.
Проект аннотации-вкладыша подается согласно национальной форме на румынском языке.
Маркировка упаковки также подается на румынском. Дополнительные языки разрешены при условии идентичности информации с румынским.
Для лекарственных средств, предназначенных к применению медицинским персоналом, например: госпитального назначения (лекарственные средства для анестезии, растворы для инфузий, вакцины, радиофармацевтические препараты, производные крови или сыворотки крови), или “препаратов-сирот” или лекарственных препаратов, применяемых в целях заместительной терапии, допускается предоставление первичной и вторичной упаковки на языке/языках международного значения.
Внесение изменений
Владелец регистрационного свидетельства в течение периода его действия обязан извещать и получать разрешение Агентства по лекарствам и медицинским изделиям относительно любых изменений.
Типы изменений в Молдове приближены к требованиям ЕС:
- Срочное ограничение для безопасности;
- Изменение содержания (терминологии);
- Изменения типа IА;
- Изменения типа IБ;
- Изменения типа II;
- Передача Регистрационного свидетельства;
- Расширение линии (изменения, которые ведут к выдаче нового регистрационного свидетельства).
Каждое изменение подается отдельным Заявлением, за исключением последовательных изменений (изменение, которое является неизбежным результатом другого изменения).
Утвержденные изменения должны быть приведены в исполнение в срок не более 6 месяцев с даты их утверждения. Владелец регистрационного свидетельства обязан проинформировать Агентство по лекарствам и медицинским изделиям о дате введения серий в оборот в соответствии с новыми утвержденными пострегистрационными изменениями.
Перерегистрация (повторная авторизация)
В срок не более чем за 6 месяцев до окончания срока действия регистрационного свидетельства Заявитель может подать Заявление в Агентство по Лекарствам и медицинским изделиям на повторную авторизацию (перерегистрацию).
Список документации, необходимой для повторной авторизации, уменьшен по сравнению с новой регистрацией. В ходе перерегистрации Заявитель должен подтвердить отсутствие изменений, либо подать их на утверждение.
В ходе перерегистрации может потребоваться лабораторный контроль качества образцов. Такой контроль не проводится при условии, если отсутствуют изменения в спецификации на готовое лекарственное средство, и за последние 5 лет ни разу не был получен отказ в импорте.
Фармаконадзор
Система фармаконадзора в Молдове построена с учетом норм, применяемых в международной практике, а именно — Руководства Международной конференции по гармонизации технических требований к регистрации лекарственных средств для человека (ICH) и Директивы Совета Европейского Экономического Сообщества по вопросам фармакологического надзора №75/319.
Компетентным органом Молдовы, осуществляющим надзор за нежелательными явлениями, регистрацию, систематизацию и анализ данных о побочных реакциях, является Агентство по лекарственным и медицинским изделиям.
Информация о побочных реакциях/действиях лекарственных средств поступает в Отдел регистрации лекарств, клинической оценки и фармаконадзора от:
- врачей и фармацевтов всех медицинских учреждений Республики Молдова, независимо от форм собственности;
- производителей/владельцев регистрационного свидетельства или их уполномоченных представителей;
- уполномоченных международных организаций (ВОЗ – Всемирная Организация Здравоохранения, ЕС – Европейское сообщество и т. п.);
- медицинских информационных источников и научных изданий;
- общественных организаций, которые представляют интересы потребителей лекарственных средств, а также граждан;
- комиссии по вопросам этики (во время клинических испытаний лекарственных средств);
- исследователей клинических баз, вовлеченных в проведение клинических испытаний лекарственных средств.
Согласно требованиям законодательства, Заявитель должен назначить Уполномоченное лицо в Республике Молдова для осуществления фармаконадзора. К Заявлению на регистрацию прилагается curriculum vitae такого уполномоченного лица.
Для изменения такого уполномоченного лица потребуется подавать Заявление на проведение соответствующих изменений.
Производители/ владельцы регистрационного свидетельства (или их уполномоченные представители) лекарственного средства, разрешенного к медицинскому применению, обязаны предоставлять Отделу фармаконадзора информацию о любом побочном действии лекарственного средства, на протяжении первых 5 лет после получения регистрационного свидетельства.
Сообщение о непредвиденной серьезной нежелательной реакции на лекарственное средство, выявленной на территории Молдовы и других стран подается в течении 15 дней с момента получения информации.
В течение периода действия регистрационного свидетельства лекарственного средства Владелец подает в Агентство по лекарствам и медицинским изделиям периодические отчеты о безопасности (PSUR) зарегистрированного лекарственного средства, которые представляются со следующей периодичностью:
- один раз в 6 месяцев в течение первых 2 лет размещения лекарственного препарата на рынке;
- один раз в год в течение следующих 3 лет;
- впоследствии отчеты представляются с интервалом в 5 лет: на этапе следующего получения авторизации (фактически — при перерегистрации).
Агентство по лекарствам и медицинским изделиям может потребовать проведение проверки у владельца регистрационного свидетельства или его представительства для проверки соблюдения требований и соответствия системе фармаконадзора с завершением отчета о проверке в срок до 30 рабочих дней и его представлением на заседании Комиссии по лекарствам.
Изделия медицинского назначения
Все медицинские изделия, до размещения на рынке Молдовы, подлежат обязательной маркетинговой авторизации, которая выполняется:
- если медицинское изделие имеет СЕ-маркировку: путем уведомления,
- либо, если медицинское изделие не имеет СЕ-маркировки: путем национальной оценки соответствия и регистрации.
Медицинские изделия, допущенные на рынок ЕС (имеющие СЕ-маркировку) проходят упрощенную процедуру допуска на рынок путем уведомления (нотификации). Законодательство определяет, что так как такое изделия уже прошли оценку соответствия, то нет необходимости повторения данных процедур.
Изделие размещается на рынке без нанесения национального знака соответствия SM, однако до размещения изделия на рынке необходимо:
- Назначить Уполномоченного представителя в Молдове, для чего производитель должен доверенностью или Договором передать необходимые права юридическому лицу-резиденту Молдовы;
- Провести регистрацию медицинского изделия путем уведомления (нотификации) Агентства по лекарственным и медицинским изделиям;
- В случае, если медицинское изделие попадает под действие других технических регламентов — убедиться, что выполнены все требования таких регламентов;
- Выполнить необходимые законодательные требования к маркировке и инструкции по применению (руководству пользователя).
Медицинские изделия, которые не были ранее допущены на рынок ЕС, должны пройти процедуру оценки соответствия согласно законодательству Молдовы, а затем — внесены в Государственный реестр медицинских изделий. Для этого необходимо:
- Назначить Уполномоченного представителя в Молдове, для чего производитель должен доверенностью или Договором передать необходимые права юридическому лицу-резиденту Молдовы;
- Выполнить необходимые законодательные требования к маркировке и инструкции по применению (руководству пользователя).
- В зависимости от класса медицинского изделия:
- Для I-го класса (нестерильного, без функций измерения) — составить Техническую документацию и выдать Декларацию соответствия требованиям Положения об установлении условий размещения на рынке медицинских изделий.
- Для изделий других классов — выбрать процедуру оценки соответствия (на партию, с инспектированием и пр.), подать Заявку и необходимую техническую документацию в назначенный орган по оценке соответствия, пройти предусмотренные действия и получить сертификат соответствия.
- В случае, если медицинское изделие попадает под действие других технических регламентов — убедиться, что выполнены все требования таких регламентов;
- Провести регистрацию медицинского изделия путем уведомления (нотификации) Агентства по лекарственным и медицинским изделиям;
- Нанести национальный знак (марку) соответствия SM.
Уполномоченный представитель производителя
Если производитель медицинского изделия не является резидентом Республики Молдова, то такой производитель должен назначить Уполномоченного представителя. Назначение уполномоченного представителя выполняется Договором или Доверенностью. Уполномоченный представитель может получить право инициировать процедуры оценки соответствия.
Обязанности Уполномоченного представителя:
- нанести на медицинские изделия, которые прошли процедуру маркетинговой авторизации, свое наименование и адрес;
- зарегистрироваться в Агентстве по лекарственным средствам и медицинским изделиям, и предоставить описание изделий, которые являются предметом его деятельности для внесения данных в базу Агентства;
- получать данные об инцидентах от Агентства и предпринимать необходимые действия;
- предпринимать необходимые действия в случае обнаружения медицинских изделий без надлежащей маркировки;
- хранить документацию и предоставить к ней доступ для Агентства, в течении 5 лет с момента выведения на рынок медицинского изделия:
- декларацию соответствия;
- техническую документацию
- изменения в техническую документацию;
- решения и документы назначенного органа.
Маркировка упаковки и инструкция по применению (руководство пользователя)
Информация на маркировке изделия может быть указана в виде международных символов. Маркировка медицинского изделия, инструкция по применению (руководство пользователя) должны быть предоставлены пользователю и пациенту на молдавском (румынском) языке. Так, на маркировке медицинского изделия, которое содержит международные символы, на молдаврумынском языке необходимо указать:
- наименование или фирменное название производителя, наименование и адрес уполномоченного представителя в Республике Молдова;
- название медицинского изделия изделия и другие данные, необходимые для идентификации изделия и содержания упаковки;
- если изделие изготовлено на заказ, на нем следует указывать “dispozitiv fabricat la comandă”;
- если изделие предназначено для клинических исследований, на нем следует указывать “exclusiv pentru investigaţii clinice”;
- любые специальные условия хранения и/или обращения;
- любые специальные рабочие инструкции;
- любые предупреждения и/или меры предосторожности, которые необходимо принимать;
- если применимо — информацию о том, что изделие содержит вещества, производные из крови человека.
Национальная марка соответствия SM (Securitatea conform cerinţelor esenţiale Moldova) указывает, что производитель или его Уполномоченный представитель несет ответственность за нанесение данной марки, проверил соответствие продукции всем основным требованиям, применяемым в технических регламентах, и что данная продукция подвергалась процедурам оценки соответствия, предусмотренным всеми применимыми техническими регламентами.
|
|
|
|
Графическое изображение знака SM |
Технические требования (пропорции) к знаку SM |

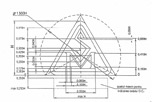
За национальным знаком соответствия должен следовать идентификационный номер назначенного органа, если указанный орган участвовал в процедуре оценки соответствия. Идентификационный номер органа наносится самим органом либо, по указанию такового, производителем или его уполномоченным представителем.
Специальные пищевые продукты
Добавки к пище могут быть размещены на рынке Республики Молдова только при условии их соответствия требованиям Санитарного регламента о добавках к пище. До размещения на рынке БАДы подлежат процедурам нотификации (витамины и/или минералы) или регистрации (БАДы, содержащие другие вещества).
Регистрацию специальных пищевых продуктов проводит Министерство здравоохранения Республики Молдова на основании результатов оценки и отчета Национального центра общественного здоровья.
Для нотификации в Национальный центр общественного здоровья подается формуляр нотификации с копией этикетки (в оригинале и с переводом на румынский язык).
Для регистрации подается Заявление и досье, согласно рекомендациям, утвержденным Министерством здравоохранения. Национальный центр общественного здоровья проводит оценку документов и представляет отчет с рекомендациями по регистрации продукта Министерству здравоохранения, которое издает приказ о регистрации (или отказе в регистрации) с включением в «Список нотифицированных/зарегистрированных пищевых добавок».
Маркировка этикетки и инструкции по применению выполняется на румынском языке.
Косметическая продукция
Косметическая продукция на стадии импорта проходит сертификацию. Предварительная регистрация косметической продукции не требуется.
Маркировка этикетки выполняется на румынском языке.
Компания «Кратия» предлагает выполнение работ по государственной регистрации лекарственных средств, созданию и поддержанию системы фармаконадзора, маркетинговой авторизации (нотификации, оценки соответствия, регистрации) изделий медицинского назначения, регистрации специальных пищевых продуктов в Молдове.
Для начала сотрудничества или получения консультации Вы можете связаться с нами:
- по телефонам +38 044 361-48-28, +38 044 221-71-29,
- по e-mail info@cratia.ua,
- или приехать на встречу к нам в офис.