Молдова
Населення Молдови становить близько 3,5 млн. чоловік. Ринок медичної і фармацевтичної продукції на більш ніж 80% забезпечується за рахунок імпорту. Обсяг ринку лікарських засобів і медичних виробів щороку зростає. У 2016-2017 роках уряд Молдови вдався до низки дій, спрямованих на збільшення кількості реімбурсованих лікарських засобів, залучення міжнародних виробників і поліпшення конкуренції на фармацевтичному ринку, а також розширює мережі аптек у сільській місцевості.
Регуляторна система Республіки Молдови є незалежною, однак законодавство щодо медичної продукції знаходиться в активній гармонізації із законодавством Європейського Союзу. Спрощені процедури реєстрації передбачені для медичних продуктів, які отримали авторизацію на ринку ЄС та інших країн з високими регуляторними вимогами.
Для допуску на ринок Молдови лікарських засобів необхідно провести державну реєстрацію. Усі медичні вироби підлягають державній реєстрації, заснованій на процедурі оцінки відповідності, до їх розміщення на ринку Молдови. Спеціальні харчові продукти проходять процедуру нотифікації або реєстрації, і повинні відповідати вимогам Санітарного регламенту про добавки до їжі. Імпортовані косметичні продукти проходять сертифікацію в процесі митного оформлення.
Лікарські засоби
Основними законодавчими актами, що регламентують процедуру реєстрації та обігу лікарських засобів в Молдові є:
- Закон Республіки Молдова №1456-XII від 25.05.1993 «Про фармацевтичну діяльність»;
- Закон Республіки Молдова №1409-XIII від 17.12.1997 «Про ліки»;
- Наказ Міністерства охорони здоров’я Республіки Молдова №739 від 23.07.2012 «Про авторизацію лікарських засобів для людини та затвердження постреєстраційних змін».
Реєстрацію лікарських засобів координує Міністерство охорони здоров’я Республіки Молдова, а експертизу реєстраційних матеріалів проводить Агентство з лікарських і медичних виробів (https://msmps.gov.md/ru/).
Заявник реєстрації – особа, призначена та уповноважена власником в якості його представника під час проведення процедури авторизації лікарських засобів в Республіці Молдова. Власник реєстраційного свідоцтва лікарського засобу – винахідник, виробник або будь-яка інша юридична особа, уповноважена ними, відповідальна за ефективність, якість та безпеку лікарського засобу. Заявником реєстрації може бути тільки резидент Республіки Молдова, в той час як власником може бути як резидент, так і нерезидент.
Власник реєстраційного свідоцтва несе відповідальність за якість, ефективність та безпечність лікарського засобу, розміщеного на ринку, і його відповідність вимогам Специфікації.
Лікарський засіб, що подається на первинну авторизацію в Республіці Молдова, повинен бути зареєстрований або в країні виробництва, або в країні власника реєстраційного свідоцтва, або в одній з країн Європейського Економічного Простору, Швейцарії, США, Канаді, Японії, Австралії.
Процедура реєстрації складається з наступних етапів:
- Заявник подає до Агентства з ліків і медичних виробів формуляр заяви, а також – реєстраційне досьє, зразки лікарського засобу та референтні стандарти.
- Агентство з ліків і медичних виробів виставляє Заявнику рахунки для оплати суми за реєстрацію.
- Після надходження оплати проводиться первинна експертиза: перевіряється комплектність досьє. При позитивному результаті первинної експертизи заява валідується, при негативному результаті – досьє повертається заявнику.
- Валідована заява на реєстрацію і документація для реєстрації розподіляються групі експертів і за необхідності, лабораторії з контролю якості ліків.
- Проводиться спеціалізована експертиза реєстраційного досьє.
- Лабораторія з контролю якості ліків проводить апробацію зразків лікарського засобу відповідно до Специфікації і методів контролю.
- Під час спеціалізованої експертизи та/або лабораторного аналізу зразків можуть виникнути зауваження, на які Заявнику необхідно надати повну відповідь на протягом не більше 90 днів. Термін може бути збільшений письмовою заявою заявника не більше ніж на 30 днів.
- У процесі експертизи може бути прийнято рішення про проведення перевірки виробництва, місць проведення доклінічних/клінічних випробувань, а також перевірки у власника реєстраційного посвідчення або його представництва відносно відповідності системи фармаконагляду. На час такої перевірки (інспекції) процедура реєстрації припиняється, але не більше ніж на 12 місяців.
- Позитивні звіти з спеціалізованої експертизи і лабораторного контролю якості представляються Комісії з ліків, яка приймає рішення про затвердження лікарських засобів до реєстрації.
- Агентство з ліків і медичних виробів складає проект наказу про авторизацію лікарських засобів, який потім затверджується Міністерством охорони здоров’я.
- Лікарські засоби, внесені до наказу Міністерства охорони здоров’я, вважаються включеними до Державного реєстру ліків, дозволених в Республіці Молдова, з дня підписання відповідного наказу.
- Після підписання наказу видається оригінал реєстраційного свідоцтва.
Термін спеціалізованої експертизи становить до 210 днів. Однак даний термін не включає в себе (а) дії, які виконуються до початку експертизи: отримання і оплата рахунків, первинна експертиза, (б) час, необхідний заявникові для підготовки відповідей на зауваження, (в) час, необхідний для проведення перевірки виробництва, місць проведення доклініки/клініки, місця ведення фармаконагляду, (г) дії, які виконуються після прийняття рішення про затвердження лікарського засобу до реєстрації: складання проекту Наказу та його підписання, включення ліків до реєстру (регістру), видача оригіналу свідоцтва.
Заявник має право призупинити процес авторизації на термін до 5 років.
Лабораторний контроль якості не проводиться:
- для лікарських засобів, зареєстрованих EMA або вироблених в одній з країн Європейського економічного простору або в Швейцарії, США, Канаді, Японії, Австралії;
- для лікарських засобів, місце виробництва яких було інспектовано однією з країн-учасниць/членом PIC/S, підтверджено сертифікатом GMP, виданих уповноваженим органом цієї країни.
Термін дії реєстраційного свідоцтва становить 5 років. Лікарський засіб може використовуватися в медичній практиці до закінчення терміну придатності.
Реєстраційне свідоцтво може бути анульовано Міністерством охорони здоров’я в разі, якщо лікарський засіб не поставлявся в країну протягом трьох (3) років з моменту видачі реєстраційного посвідчення.
Спрощена процедура реєстрації
Спрощена процедура реєстрації передбачена законодавством Молдови для лікарських засобів, зареєстрованих EMA як мінімум в одній з країн Європейського економічного простору або в Швейцарії, США, Канаді, Японії, Австралії.
Очевидними перевагами спрощеної процедури є зменшений термін експертизи до 60 днів, відсутність необхідності надання зразків і стандартів, відсутність лабораторного контролю якості.
Спрощена процедура реєстрації діє за умови повної ідентичності поданого досьє останньому (актуальному) прийнятому досьє в одній із зазначених країн. За наявності досьє, специфічного для ринку або регіону препарат повинен подаватися за стандартною процедурою.
Вимоги до реєстраційного досьє
Реєстраційне досьє подається в форматі CTD:
- Модуль 1 подається в паперовому форматі. Частина адміністративної документації підлягає легалізації. Eudra сертифікати дозволено подавати без нотаріального засвідчення.
- Модулі 2-5 подаються на електронному носії.
Документація подається або румунською, або англійською, або російською мовою.
SmPC подається в перекладі на румунськоюу мовоюу.
Проект анотації-вкладиша подається згідно з національною формою румунською мовою.
Маркування упаковки також подається румунською. Додаткові мови дозволені за умови ідентичності інформації з румунською.
Для лікарських засобів, призначених для застосування медичним персоналом, наприклад: госпітального призначення (лікарські засоби для анестезії, розчини для інфузій, вакцини, радіофармацевтичні препарати, похідні крові або сироватки крові), або “препаратів-сиріт” або лікарських препаратів, що застосовуються в цілях замісної терапії, допускається надання первинної та вторинної упаковки мовою/мовами міжнародного значення.
Внесення змін
Власник реєстраційного свідоцтва протягом періоду його дії зобов’язаний сповіщати і отримувати дозвіл Агентства з ліків і медичних виробів щодо будь-яких змін.
Типи змін в Молдові наближені до вимог ЄС:
- Термінове обмеження для безпеки;
- Зміна змісту (термінології);
- Зміни типу IА;
- Зміни типу IБ;
- Зміни типу II;
- Передача Реєстраційного свідоцтва;
- Розширення лінії (зміни, які ведуть до видачі нового реєстраційного свідоцтва).
Кожна зміна подається окремою Заявою, за винятком послідовних змін (зміна, яка є неминучим результатом іншої зміни).
Затверджені зміни повинні бути приведені у виконання в строк не більше 6 місяців з дати їх затвердження. Власник реєстраційного свідоцтва зобов’язаний проінформувати Агентство з ліків і медичних виробів про дату введення серій в оборот відповідно до нових затверджених післяреєстраційних змін.
Перереєстрація (повторна авторизація)
В термін не більше ніж за 6 місяців до закінчення терміну дії реєстраційного посвідчення Заявник може подати Заяву в Агентство з Ліків і медичних виробів на повторну авторизацію (перереєстрацію).
Список документації, необхідної для повторної авторизації, зменшений в порівнянні з новою реєстрацією. Під час перереєстрації Заявник повинен підтвердити відсутність змін, або подати їх на затвердження.
Під час перереєстрації може знадобитися лабораторний контроль якості зразків. Такий контроль не проводиться за умови, якщо відсутні зміни в специфікації на готовий лікарський засіб, і за останні 5 років жодного разу не було отримано відмови в імпорті.
Фармаконагляд
Система фармаконагляду в Молдові побудована з урахуванням норм, що застосовуються в міжнародній практиці, а саме – Керівництва Міжнародної конференції з гармонізації технічних вимог до реєстрації лікарських засобів для людини (ICH) і Директиви Ради Європейського Економічного Співтовариства з питань фармакологічного нагляду №75/319.
Компетентним органом Молдови, який здійснює нагляд за небажаними явищами, реєстрацію, систематизацію та аналіз даних про побічні реакції, є Агентство з лікарських і медичних виробів.
Інформація про побічні реакції/дії лікарських засобів надходить до Відділу реєстрації ліків, клінічної оцінки та фармаконагляду від:
- лікарів і фармацевтів усіх медичних установ Республіки Молдова, незалежно від форм власності;
- виробників/власників реєстраційного свідоцтва або їх уповноважених представників;
- уповноважених міжнародних організацій (ВООЗ – Всесвітня організація охорони здоров’я, ЄС – Європейське співтовариство і т. п.);
- медичних інформаційних джерел та наукових видань;
- громадських організацій, які представляють інтереси споживачів лікарських засобів, а також громадян;
- комісії з питань етики (під час клінічних випробувань лікарських засобів);
- дослідників клінічних баз, залучених до проведення клінічних випробувань лікарських засобів.
Згідно з вимогами законодавства, Заявник повинен призначити Уповноважену особу в Республіці Молдова для здійснення фармаконагляду. До Заяви на реєстрацію додається curriculum vitae такої уповноваженої особи.
Для зміни такої уповноваженої особи потрібно подавати Заяву на проведення відповідних змін.
Виробники/власники реєстраційного свідоцтва (або їх уповноважені представники) лікарського засобу, дозволеного до медичного застосування, зобов’язані надавати Відділу фармаконагляду інформацію про будь-яку побічну дію лікарського засобу, протягом перших 5 років після отримання реєстраційного свідоцтва.
Повідомлення про непередбачену серйозну небажану реакцію на лікарський засіб, виявлену на території Молдови та інших країн подається протягом 15 днів з моменту отримання інформації.
Протягом періоду дії реєстраційного свідоцтва лікарського засобу Власник подає до Агентства з ліків і медичних виробів періодичні звіти про безпеку (PSUR) зареєстрованого лікарського засобу, які представляються з такою періодичністю:
- один раз на 6 місяців протягом перших 2 років розміщення лікарського препарату на ринку;
- один раз на рік протягом наступних 3 років;
- згодом звіти подаються з інтервалом в 5 років: на етапі наступного отримання авторизації (фактично – при перереєстрації).
Агентство з ліків і медичних виробів може потребувати проведення перевірки у власника реєстраційного посвідчення або його представництва для перевірки дотримання вимог і відповідності системі фармаконагляду із завершенням звіту про перевірку в термін до 30 робочих днів і його поданням на засіданні Комісії з ліків.
Вироби медичного призначення
Усі медичні вироби, до розміщення на ринку Молдови, підлягають обов’язковій маркетинговій авторизації, яка виконується:
- якщо медичний виріб має СЕ-маркування: шляхом повідомлення,
- або, якщо медичний виріб не має СЕ-маркування: шляхом національної оцінки відповідності та реєстрації.
Медичні вироби, допущені на ринок ЄС (що мають СЕ-маркування) проходять спрощену процедуру допуску на ринок шляхом повідомлення (нотифікації). Законодавство визначає, що так як такі вироби вже пройшли оцінку відповідності, то немає необхідності повторення даних процедур.
Виріб розміщується на ринку без нанесення національного знака відповідності SM, проте до розміщення виробу на ринку необхідно:
- Призначити Уповноваженого представника в Молдові, для чого виробник повинен довіреністю або Договором передати необхідні права юридичній особі-резиденту Молдови;
- Провести реєстрацію медичного виробу шляхом повідомлення (нотифікації) Агентства з лікарських і медичних виробів;
- У разі, якщо медичний виріб потрапляє під дію інших технічних регламентів – переконатися, що виконані вусі вимоги таких регламентів;
- Виконати необхідні законодавчі вимоги до маркування та інструкції із застосування (керівництва користувача).
Медичні вироби, що не були раніше допущені на ринок ЄС, повинні пройти процедуру оцінки відповідності згідно із законодавством Молдови, а потім – внесені до Державного реєстру медичних виробів. Для цього необхідно:
- Призначити Уповноваженого представника в Молдові, для чого виробник повинен довіреністю або Договором передати необхідні права юридичній особі-резиденту Молдови;
- Виконати необхідні законодавчі вимоги до маркування та інструкції для застосування (керівництву користувача).
- Залежно від класу медичного виробу:
- Для I-го класу (нестерильного, без функцій вимірювання) – скласти Технічну документацію і видати Декларацію відповідності вимогам Положення про встановлення умов розміщення на ринку медичних виробів.
- Для виробів інших класів – обрати процедуру оцінки відповідності (на партію, з інспектуванням та ін.), подати Заявку та необхідну технічну документацію в призначений орган з оцінки відповідності, пройти передбачені дії і отримати сертифікат відповідності.
- У разі, якщо медичний виріб потрапляє під дію інших технічних регламентів – переконатися, що виконані всі вимоги таких регламентів;
- Провести реєстрацію медичного виробу шляхом повідомлення (нотифікації) Агентства з лікарських і медичних виробів;
- Нанести національний знак (марку) відповідності SM.
Уповноважений представник виробника
Якщо виробник медичного виробу не є резидентом Республіки Молдова, то такий виробник повинен призначити Уповноваженого представника. Призначення уповноваженого представника виконується Договором або Довіреністю. Уповноважений представник може отримати право ініціювати процедури оцінки відповідності.
Обов’язки Уповноваженого представника:
- нанести на медичні вироби, які пройшли процедуру маркетингової авторизації, своє найменування та адресу;
- зареєструватися в Агентстві з лікарських засобів і медичних виробів, і надати опис виробів, які є предметом його діяльності для внесення даних в базу Агентства;
- отримувати дані про інциденти від Агентства і вживати необхідні дії;
- вживати необхідні дії у разі виявлення медичних виробів без належного маркування;
- зберігати документацію і надати до неї доступ для Агентства, протягом 5 років з моменту виведення на ринок медичного виробу:
- декларацію відповідності;
- технічну документацію;
- зміни до технічної документації;
- рішення і документи призначеного органу.
Маркування упаковки і інструкція із застосування (керівництво користувача)
Інформація на маркуванні виробу може бути вказана у вигляді міжнародних символів. Маркування медичного виробу, інструкція дляіз застосування (керівництво користувача) повинні бути надані користувачеві і пацієнту молдавською (румунською) мовою. Так, на маркуванні медичного виробу, яке містить міжнародні символи, румунською мовою необхідно вказати:
- найменування або назву фірми виробника, найменування та адресу уповноваженого представника в Республіці Молдова;
- назву медичного виробу та інші дані, необхідні для ідентифікації виробу і змісту упаковки;
- якщо виріб виготовлений на замовлення, на ньому слід вказувати “dispozitiv fabricat la comandă”;
- якщо виріб призначений для клінічних досліджень, на ньому слід вказувати “exclusiv pentru investigaţii clinice”;
- будь-які спеціальні умови зберігання та/або поводження;
- будь-які спеціальні робочі інструкції;
- будь-які попередження та/або запобіжні заходи, які необхідно приймати;
- якщо може бути застосовано – інформацію про те, що виріб містить речовини, похідні з крові людини.
Національна марка відповідності SM (Securitatea conform cerinţelor esenţiale Moldova) вказує, що виробник або його Уповноважений представник несе відповідальність за нанесення даної марки, перевірив відповідність продукції усім основним вимогам, що застосовуються в технічних регламентах, і що дана продукція піддавалася процедурам оцінки відповідності, передбаченим усіма застосовними технічними регламентами.
|
|
|
|
Графічне зображення знака SM |
Технічні вимоги (пропорції) до знаку SM |

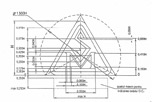
За національним знаком відповідності повинен слідувати ідентифікаційний номер призначеного органу, якщо зазначений орган брав участь в процедурі оцінки відповідності. Ідентифікаційний номер органу наноситься самим органом або, за вказівкою такого, виробником або його уповноваженим представником.
Спеціальні харчові продукти
Добавки до їжі можуть бути розміщені на ринку Республіки Молдова тільки за умови їх відповідності вимогам Санітарного регламенту про добавки до їжі. До розміщення на ринку БАДи підлягають процедурам нотифікації (вітаміни та/або мінерали) або реєстрації (БАДи, що містять інші речовини).
Реєстрацію спеціальних харчових продуктів проводить Міністерство охорони здоров’я Республіки Молдова на підставі результатів оцінки та звіту Національного центру громадського здоров’я.
Для нотифікації в Національний центр громадського здоров’я подається формуляр нотифікації з копією етикетки (в оригіналі із перекладом на румунською мовою).
Для реєстрації подається Заява та досьє, відповідно до рекомендацій, затверджених Міністерством охорони здоров’я. Національний центр громадського здоров’я проводить оцінку документів і подає звіт з рекомендаціями по реєстрації продукту Міністерству охорони здоров’я, яке видає наказ про реєстрацію (або відмову в реєстрації) з включенням в «Список нотифікованих/зареєстрованих харчових добавок».
Маркування етикетки та інструкції із застосування виконується румунською мовою.
Косметична продукція
Косметична продукція на стадії імпорту проходить сертифікацію. Попередня реєстрація косметичної продукції не потребується.
Маркування етикетки виконується румунською мовою.
Компанія «Кратія» пропонує виконання робіт з державної реєстрації лікарських засобів, створення і підтримки системи фармаконагляду, маркетингової авторизації (нотифікації, оцінки відповідності, реєстрації) виробів медичного призначення, реєстрації спеціальних харчових продуктів в Молдові.
Для початку співпраці або отримання консультації Ви можете зв’язатися з нами:
- за телефонами +38 044 361-48-28, +38 044 221-71-29,
- по e-mail: info@cratia.ua,
- або приїхати на зустріч до нас в офіс .